This vignette will show the most suitable commands to work with pulse-chase data.
Throughout this vignette, we will be using the GRAND-SLAM processed SLAM-seq data set from Herzog et al. 2017 [1]. The data set includes SLAM-seq pulse-chase samples of mouse embryonic stem cells (mESCs) with pulse durations of 0, 0.5, 1, 3, 6, 12, and 24 hours.
suppressPackageStartupMessages({
library(grandR)
library(ggplot2)
library(patchwork)
options(timeout = 600)
})First we load the data and do the standard preprocessing steps. For more on these initial commands see the Loading data and working with grandR objects vignette.
In this step, we need to modify the sample names in the data, as the
names were not processed using systematic naming conventions. If
systematic sample names were used, the rename.sample
parameter would not be necessary. This parameter allows us to specify a
function for renaming the samples. The specified function removes all
occurrences of “.chase” and replaces 0.5 with 0_5. For example,
“mESC.0.5h.chase.B” becomes “mESC.0_5h.B” and “mESC.0h.chase.C” becomes
“mESC.0h.C”. The output of the function is the modified vector after
these replacements. This enables us to load the data using the design
vector.
d=ReadGRAND("https://zenodo.org/record/7612564/files/chase_notrescued.tsv.gz?download=1",
design=c(NA,"Time",Design$Replicate),
rename.sample=function(v)
gsub(".chase","",gsub("0.5h","0_5h",v))
)
d=FilterGenes(d)
d=Normalize(d)The FitKinetics function can be utilized to analyze
pulse-chase experiment data and estimate the synthesis and degradation
rates of genes. To perform this analysis, the type parameter of the
function should be set to “chase”. The function requires a grandR object
as input, which holds the data from the pulse-chase experiment.
In addition to the required input, the FitKinetics
function also has several optional parameters that allow for
customization of the analysis. For further information on these
parameters, please refer to the Kinetic
modeling vignette.
The FitKinetics function uses the information in the
grandR object and the specified parameters to fit a non-linear least
squares model that describe the pulse-chase data. After fitting the
models, the function returns a grandR object with an added analysis
table that contains the inferred synthesis and degradation rates.
SetParallel()NULL
d=FitKinetics(d,time="Time",type="chase")The function GetAnalysisTable can be used to retrieve
the results from this analysis.
head(GetAnalysisTable(d)) Gene Symbol Length Type kinetics.Synthesis
Qsox1.155778412 Qsox1.155778412 Qsox1.155778412 258 Unknown 27.46546
Ipo9.135384724 Ipo9.135384724 Ipo9.135384724 250 Unknown 42.07806
Rpl37a.72713813 Rpl37a.72713813 Rpl37a.72713813 437 Unknown 180.72132
Igfbp2.72852476 Igfbp2.72852476 Igfbp2.72852476 255 Unknown 210.64554
Plekhb2.34879585 Plekhb2.34879585 Plekhb2.34879585 258 Unknown 36.78982
Gmppa.75443176 Gmppa.75443176 Gmppa.75443176 252 Unknown 11.25888
kinetics.Half-life
Qsox1.155778412 3.450422
Ipo9.135384724 6.117658
Rpl37a.72713813 10.772538
Igfbp2.72852476 8.569694
Plekhb2.34879585 2.462591
Gmppa.75443176 6.073079We can also visualize single genes graphically (this is the same gene as used in Fig 4a in Herzog et al. 2017 [1]):
PlotGeneProgressiveTimecourse(d,gene=Genes(d,"Dnmt3b",regex=TRUE),time="Time",type="chase")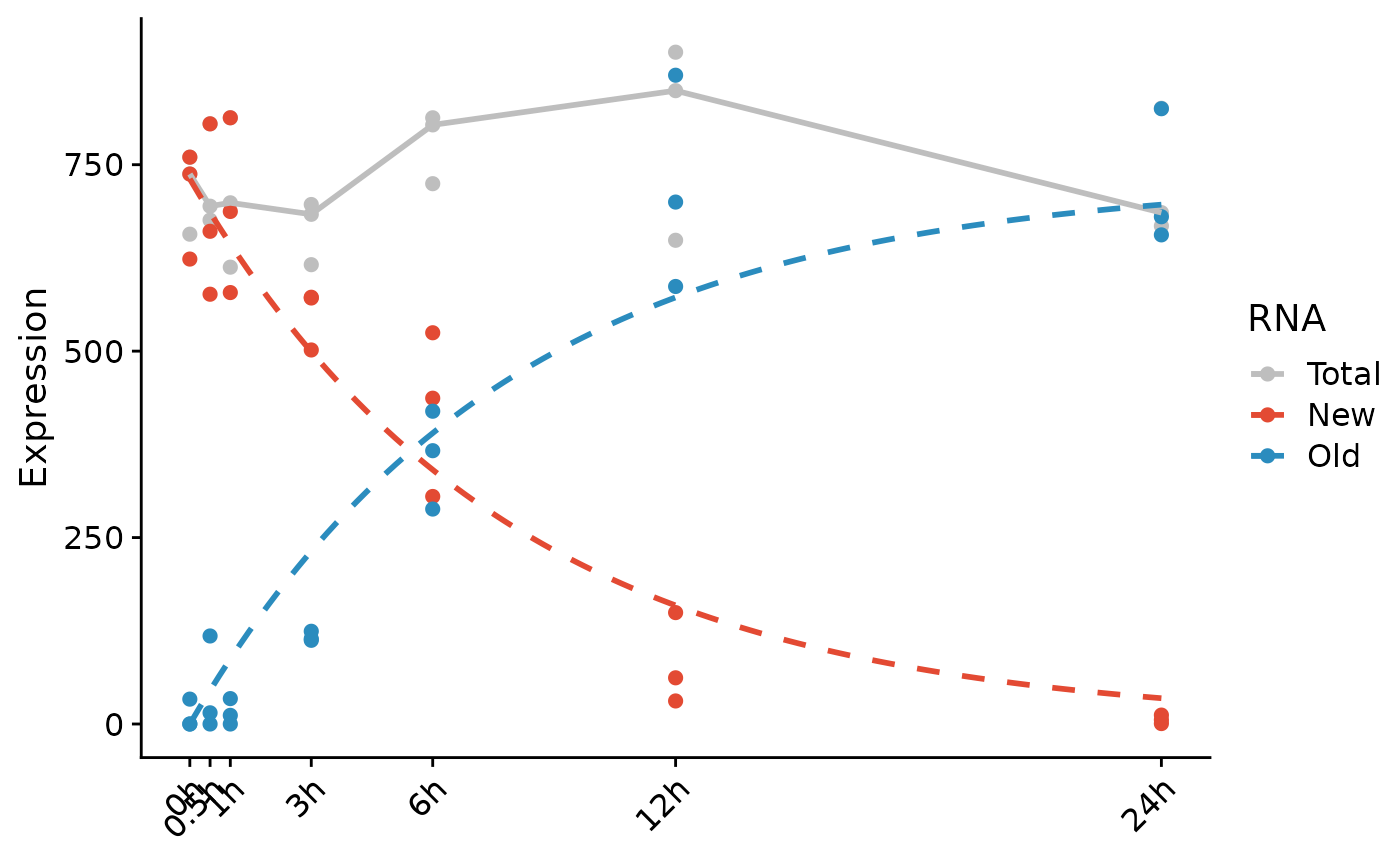
The curves represent the fitted model for this gene. Note that we do
not specify the gene name directly, since here the gene names were
labeled differently, and the Genes() function can be used
to find the matching label:
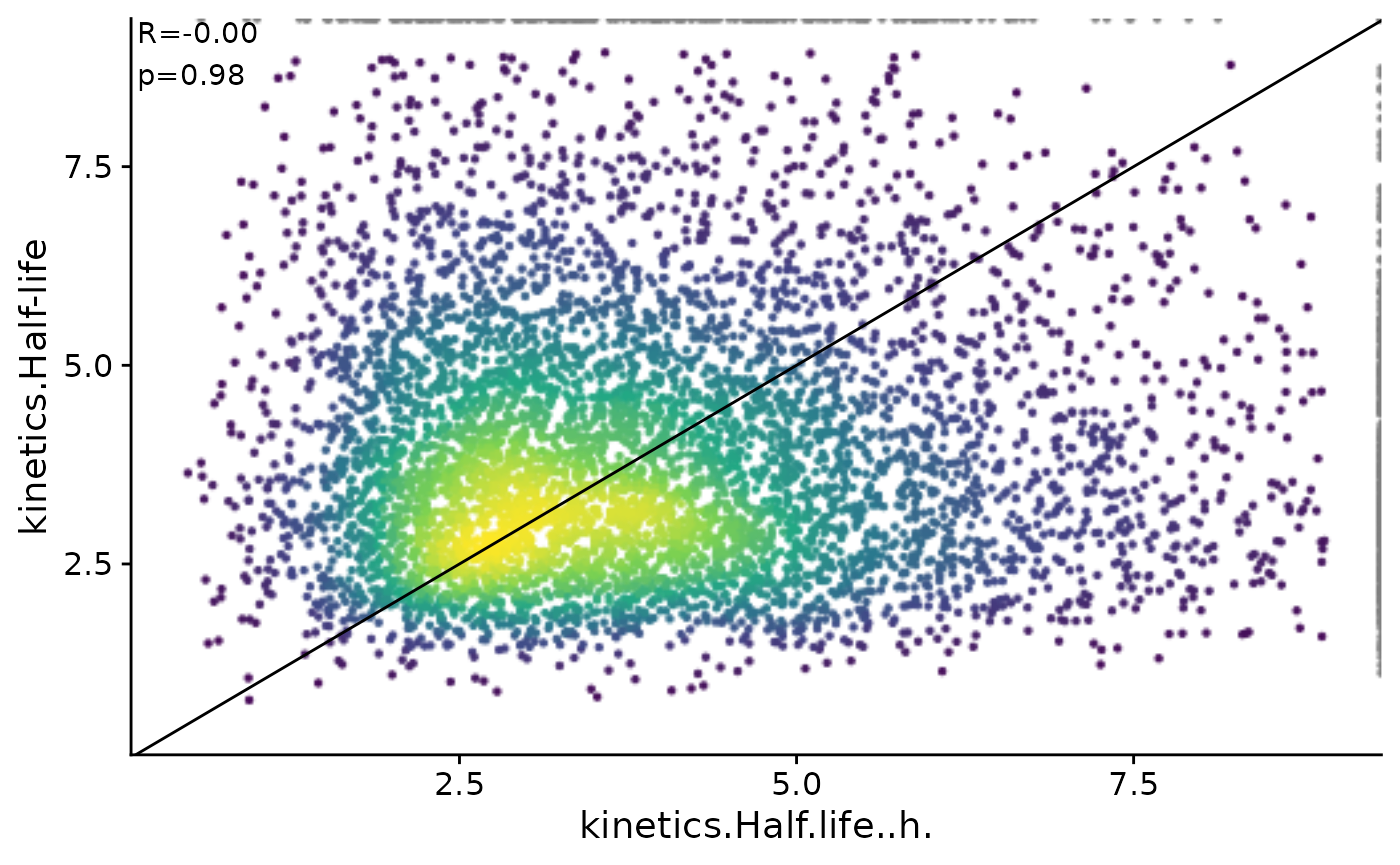
Genes(d,"Dnmt3b",regex=TRUE)[1] "Dnmt3b.153687730"In this step, we compare the half-life values obtained from the
FitKinetics function with those from Herzog et al. The
latter was estimated by observing the decrease in mismatches during the
chase period, while our method also considers the GRAND-SLAM-estimated
NTR and posterior. To compare, we add Herzog et al’s half-life values to
our analysis table and create a scatter plot with half-life values on
both the x and y axes. The plot clearly shows a correlation between the
two sets of results, indicating that the FitKinetics
results are consistent with those from Herzog et al.
t=read.delim("https://zenodo.org/record/7612564/files/halflifes.tsv?download=1")
t$Gene=paste0(t$Name,".",t$End)
d = AddAnalysis(data = d,"kinetics",table = t,by="Gene")
PlotScatter(d,`kinetics.Half.life..h.`,`kinetics.Half-life`,
correlation = FormatCorrelation())+geom_abline()